Diferencia entre alfa y talasemia beta
Diferencia clave: alfa vs talasemia beta
La talasemia es un grupo heterogéneo de trastornos causados por mutaciones hereditarias que disminuyen la síntesis de cadenas alfa o beta globina, lo que lleva a la anemia, la hipoxia tisular y la hemólisis de los glóbulos rojos relacionados con el desequilibrio en la síntesis de la cadena globina. Hay dos formas principales de talasemia como alfa talasemia y beta talassemia. En la talasemia alfa, hay una disminución en el número de cadenas de globina alfa, mientras que en la beta-talasemia es el número de cadenas de beta globina lo que se cae. Esta es la diferencia clave entre la talasemia alfa y beta.
CONTENIDO
1. Descripción general y diferencia de claves
2. Qué alfa talassemia
3. Qué beta talasemia
4. Similitudes entre la talasemia alfa y beta
5. Comparación de lado a lado - Alfa vs talasemia beta en forma tabular
6. Resumen
¿Qué es la talasemia alfa??
En la talasemia alfa, se eliminan algunos de los genes responsables de la codificación de las cadenas de globina alfa. El gen Alpha Globin generalmente tiene cuatro copias. La gravedad de la enfermedad depende del número de copias faltantes.
Hidropesía fetal
La síntesis de las cadenas de globina alfa se suprime por completo cuando faltan las cuatro copias del gen alfa globina. Dado que se requieren cadenas de globina alfa para la síntesis de hemoglobina fetal y adulta, esta condición no es compatible con la vida; Por lo tanto, la terminación del útero del embarazo ocurre si el feto se ve afectado por esta afección.
Enfermedad de HBH
Esta condición es causada por la ausencia de tres copias del gen Alpha Globin. Esto da como resultado una microcítica hipocrómica moderada a severa con esplenomegalia asociada.
Rasgos de talasemia alfa
Esto se debe a la ausencia o a la inactividad de una o dos copias del gen Alpha Globin. Aunque los rasgos de talasemia alfa no causan anemia, pueden disminuir el volumen corpuscular medio y los niveles medios de hemoglobina corpuscular al tiempo que aumentan el recuento de glóbulos rojos de más de 5.5*1012/L.
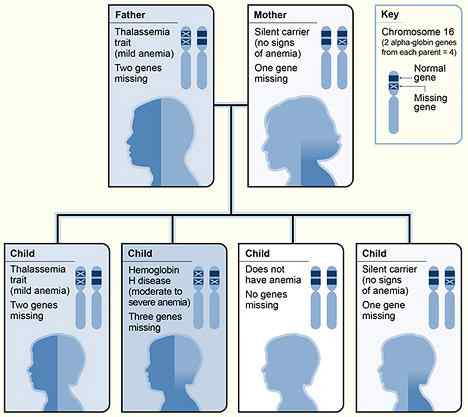
Figura 01: Herencia de talasemia alfa
El diagnóstico de talasemia alfa es a través de los estudios de síntesis de la cadena de globina.
Gestión
Los pacientes con una forma leve de anemia generalmente no requieren ningún tratamiento. La administración de hierro y ácido fólico solo se defiende en algunos pacientes. Aquellos con las formas severas de talasemia alfa requieren una transfusión de sangre de por vida.
¿Qué es la talasemia beta??
En la beta talasemia, la cantidad de cadenas de beta globina disminuye.
Beta talasemia mayor
Si ambos padres son portadores del rasgo de la beta talasemia, la posibilidad de una descendencia que herede la beta talasemia mayor es del 25%. En beta talasemia mayor, la producción de cadenas beta globina se suprime por completo o se reduce drásticamente. Dado que no hay suficientes cadenas de beta globina para que se combinen, el exceso de cadenas de globina de alfa se deposita tanto en los glóbulos rojos maduros como en los inmaduros. Esto conduce a la hemólisis prematura de los glóbulos rojos y la eritropoyesis ineficaz.
Características clínicas
- Anemia severa, que se hace evidente a los 3- 6 meses después del nacimiento.
- Esplenomegalia y hepatomegalia
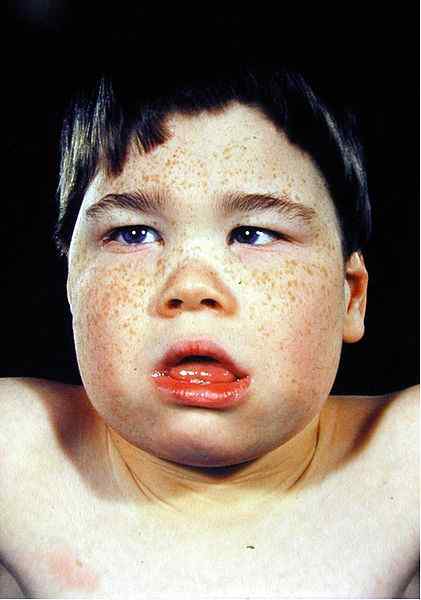
- Facies talasemáticas
Los cambios en las características faciales se deben a la expansión de los huesos debido a la hiperplasia de la médula ósea. La radiografía de rayos X muestra la apariencia de cabello sobre el cráneo que generalmente se ve en la beta talasemia.
Figura 02: facies talasemias
Diagnóstico de laboratorio
La cromatografía líquida de alto rendimiento (HPLC) es el método principal utilizado en el diagnóstico de enfermedades hematológicas hoy en día. Beta Thalassemia Mayor HPLC muestra la presencia de niveles reducidos de HBA con niveles inusualmente altos de HBF. Un recuento sanguíneo completo revelará la existencia de la anemia microcítica hipocrómica, y el examen de una película de sangre indicará la presencia de una mayor cantidad de reticulocitos junto con puntas basófilas y células objetivo.
Tratamiento
- Transfusiones de sangre regulares
- Terapia de quelación de hierro
- Ácido fólico (si la ingesta dietética del ácido fólico no es satisfactorio)
- Esplenectomía (a veces se usa para reducir el requisito de sangre)
- Trasplante de médula ósea
- Terapia génica para detección y fines terapéuticos
Rasgo de Beta Thalassemia/Menor
Beta talasemia menor es una condición común que a menudo no tiene síntomas. Aunque los signos y síntomas son similares a los de la talasemia alfa, la talasemia beta es más grave que su contraparte. El diagnóstico de beta talasemia menor se realiza si la HBA2 El nivel es más de 3.5%.
Thalassemia intermedia
Talassemia intermedia se refiere a casos de talasemia de gravedad moderada que no necesitan transfusiones regulares.
¿Cuál es la similitud entre la talasemia alfa y beta??
- En ambas condiciones, hay una disminución en el nivel de sangre de hemoglobina.
¿Cuál es la diferencia entre la talasemia alfa y beta??
Alfa vs talasemia beta | |
| Hay una disminución en el número de cadenas de globina alfa. | Hay una disminución en el número de cadenas de beta globina. |
| Eliminación de genes | |
| Se eliminan algunos de los genes responsables de la codificación de las cadenas de Alfa Globin. | Los genes responsables de la síntesis de las cadenas de beta globina se eliminan parcial o completamente. |
| Tipos | |
| Hydrops fetalis, enfermedad de HBH y rasgo de talasemia alfa son las principales formas de talasemia alfa. | Hay dos formas principales de talasemia como beta talasemia mayor y beta talasemia menor. |
| Diagnóstico | |
| El diagnóstico de talasemia alfa es a través de los estudios de síntesis de la cadena de globina. | La cromatografía líquida de alto rendimiento (HPLC) es la investigación utilizada para el diagnóstico de talasemia beta. |
| Características clínicas | |
|
|
| Tratamiento y manejo | |
|
|
Resumen -Alpha vs Beta Talassemia
La talasemia es un grupo heterogéneo de trastornos causados por mutaciones hereditarias que disminuyen la síntesis de cadenas alfa o beta globina que componen la hemoglobina hba adulta. La talasemia se puede clasificar ampliamente en dos categorías principales como alfa talasemia y talasemia beta. En la talasemia alfa, la cantidad de cadenas alfa disminuye, y en la beta-talasemia, el número de cadenas beta se reduce. Esta es la principal diferencia entre la talasemia alfa y beta.
Descargar la versión PDF de Alpha vs Beta Thalassemia
Puede descargar la versión PDF de este artículo y usarla para fines fuera de línea según la nota de cita. Descargue la versión PDF aquí Diferencia entre alfa y talasemia beta
Referencia:
1.Kumar, Parveen J., y Michael L. Aclarar. Medicina clínica de Kumar & Clark. Edimburgo: W.B. Saunders, 2009.
Imagen de cortesía:
1. "Thalassemia Alpha" del Instituto Nacional de Pulmones y Bloodos del Corazón (NIH) - Instituto Nacional del Corazón Lung and Blood (NIH) (Dominio Público) a través de Commons Wikimedia
2. "ATR-X" de Gibbons R. - Gibbons R. Alpha Thalassaemia-retraso mental, x vinculado. Orphanet j raro dis disgusto. 1, 15. 2006. doi: 10.1186/1750-1172-1-15. PMID 16722615 (CC por 2.0) a través de Commons Wikimedia