Diferencia entre ALS y MND
El diferencia clave entre ALS y MND es que MND (o enfermedad de las neuronas motoras) es una condición médica grave que causa debilidad progresiva y eventualmente La muerte debido a la insuficiencia respiratoria o la aspiración, mientras que la ELA (o la esclerosis lateral amiotrófica) es una variedad de MND con el rasgo característico del inicio gradual de debilidad en una extremidad, que se propaga a las otras extremidades y los músculos del tronco.
MND tiene cuatro variedades principales de acuerdo con los modales contrastantes de su presentación. ALS es el más común de esas cuatro variedades. Por lo tanto, ALS es simplemente una forma diferente de MND.
CONTENIDO
1. Descripción general y diferencia de claves
2. Que es ALS
3. Que es mnd
4. Similitudes entre ALS y MND
5. Comparación de lado a lado - ALS vs MND en forma tabular
6. Resumen
Que es ALS?
La esclerosis lateral amiotrófica (ELA) es la forma clínica más común de MND. Hay una presentación paraneoplásica típica, que generalmente comienza desde una extremidad, y luego se propaga gradualmente a otras extremidades y músculos del tronco. La presentación clínica es típicamente debilidad muscular focal y desperdicio, con fasciculación muscular. Los calambres también son comunes. Además, en el examen, un médico puede identificar reflejos enérgicos, respuestas plantar extensoras y espasticidad que son signos de lesiones de neuronas motoras superiores.
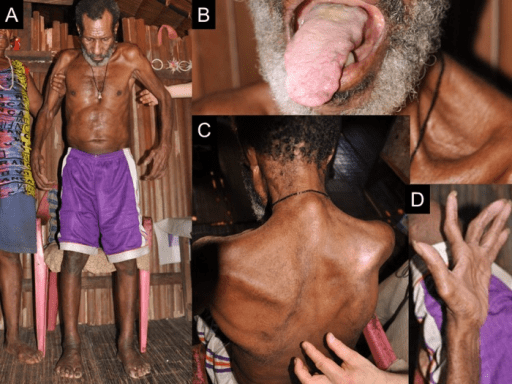
Figura 01: Imagen clínica
En casos raros, el paciente puede presentarse con paraparesia espástica asimétrica, seguido de una debilidad de tipo motor más bajo aproximadamente un mes después. El empeoramiento severo de los síntomas durante meses confirmará el diagnóstico.
Que es mnd?
MND (enfermedad de las neuronas motoras) es una condición médica grave que causa debilidad progresiva y, finalmente, la muerte debido a la insuficiencia respiratoria o la aspiración. La incidencia anual de la enfermedad es 2/100000, lo que indica que la enfermedad es relativamente infrecuente. En algunos países, los médicos identifican esta enfermedad como esclerosis lateral amiotrófica (ALS). Las personas entre 50 y 75 años suelen ser víctimas de esta enfermedad. Sin embargo, los síntomas sensoriales como el entumecimiento, el hormigueo y el dolor no ocurren en esta enfermedad, ya que no afecta al sistema sensorial.
Patogénesis
Las neuronas motoras superior e inferior en la médula espinal, los núcleos del motor del nervio craneal y las cortezas son los componentes principales del sistema nervioso central afectados por MND. Pero, otros sistemas neuronales también pueden verse afectados. Por ejemplo, en el 5% de los pacientes, se puede observar la demencia frontotemporal, mientras que en el 40% del deterioro cognitivo del lóbulo frontal de los pacientes se observa. La causa de MND es desconocida. Pero se cree ampliamente que la agregación de proteínas en los axones es la patogénesis subyacente que causa MND. La excitotoxicidad mediada por glutamato y el daño neuronal oxidativo también están involucrados en la patogénesis.
Características clínicas
Hay cuatro patrones clínicos principales, que pueden fusionarse con la progresión de la enfermedad. El más común de ellos es la esclerosis lateral amiotrófica (ELA).
Atrofia muscular progresiva
Un paciente que sufre de atrofia muscular progresiva muestra debilidad, desgaste muscular y fasciculación. Estos síntomas generalmente comienzan en una extremidad y luego se extienden a los segmentos espinales adyacentes. Esta es una presentación pura de lesión de neurona motor inferior.
Parálisis progresiva de bulbar y pseudobulbar
Los síntomas de presentación son disartria, disfagia, regurgitación nasal de fluidos y roto. Estos ocurren debido a la participación de núcleos del nervio craneal más bajos y sus conexiones supranucleares. En una parálisis mixta de bulbar, se puede observar la fasciculación de la lengua con movimientos lentos y rígidos de la lengua. Además, en la parálisis seudobulbar, se puede ver la incontinencia emocional con la risa patológica y el llanto.
Esclerosis lateral primaria
Esta es una forma rara de MND, que causa tetraparesia progresiva gradualmente y parálisis pseudobulbar.
Diagnóstico
El diagnóstico de la enfermedad se basa principalmente en la sospecha clínica. Se pueden hacer investigaciones para excluir otras causas posibles. Se puede hacer EMG para confirmar la denervación de los músculos debido a la degeneración de las neuronas motoras inferiores.
Pronóstico y manejo
No se ha demostrado que ningún tratamiento mejora el resultado. Sin embargo, el riluzol puede retrasar la progresión de la enfermedad, y puede aumentar la esperanza de vida del paciente en 3-4 meses. Además, la alimentación a través de una gastrostomía y el apoyo del ventilador no invasivo es útil para prolongar la supervivencia del paciente, aunque la supervivencia durante más de 3 años es inusual.
¿Cuáles son las similitudes entre ALS y MND??
- ALS es la variedad clínica más común de MND
- El diagnóstico de todas las formas de MND, incluida la ELA, se realiza principalmente en la sospecha clínica. EMG puede ser útil para confirmar el diagnóstico porque muestra la denervación de los músculos como resultado del daño de las neuronas motoras.
- No hay cura para ninguna forma de MND.
¿Cuál es la diferencia entre ALS y MND??
MND es una condición médica grave que causa debilidad progresiva y eventualmente la muerte debido a la insuficiencia respiratoria o la MND tiene cuatro formas principales: esclerosis lateral amiotrófica, atrofia muscular progresiva, bulbar progresivo y parálisis pseudobulbar y esclerosis lateral primaria. Para ser específico, ALS es la presentación paraneoplásica típica, que generalmente comienza desde una extremidad y luego se propaga gradualmente a otras extremidades y músculos del tronco. Esta es la diferencia clave entre ALS y MND.
Además, las características clínicas de la atrofia muscular progresiva incluyen debilidad, fasciculaciones y desgaste muscular. Estas características primero aparecen en una extremidad y luego se extienden a los segmentos espinales adyacentes. Por el contrario, la disartria, la disfagia y la regurgitación nasal de los fluidos y la redacción son las características clínicas de la parálisis progresiva de bulbar y pseudobulbar. Además, la esclerosis lateral primaria muestra tetraparesia progresiva gradualmente y parálisis pseudobulbar.

Resumen -ALS vs MND
En resumen, MND es un trastorno fatal que empeora gradualmente en última instancia, lo que resulta en la muerte cuando el paciente pierde el control sobre sus músculos respiratorios. Puede ocurrir en cuatro formas principales, de las cuales ALS es la más común. En general, esta es la diferencia entre ALS y MND.
Referencia:
1. Kumar, Parveen J., y Michael L. Aclarar. Medicina clínica de Kumar & Clark. Edimburgo: W.B. Saunders, 2009.
Imagen de cortesía:
1. "ALS Clinical Picture" de Okumiya K, Wada T, Fujisawa M, Ishine M, Garcia del Saz E, Hirata Y, Kuzuhara S, Kokubo Y, Seguchi H, Sakamoto R, Manuaba I, Watofa P, Rantetampang AL, Matsubayashi K - - - - - - - - - BMJ Open (2014) - (CC por 3.0) a través de Commons Wikimedia
2. "2213009" (CC0) a través de Pixabay


