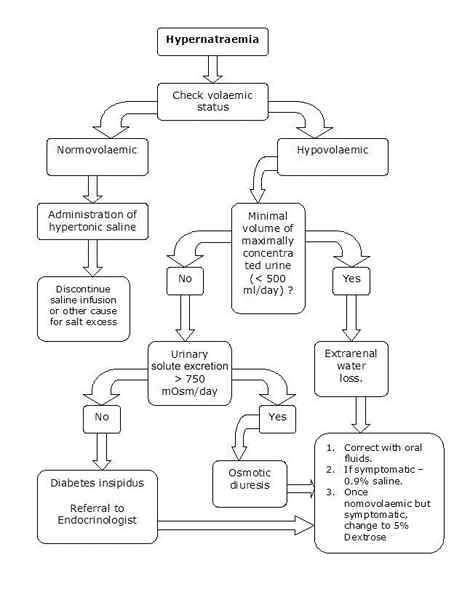
¿Cuál es la diferencia entre ALS y SMA?
El diferencia clave entre ALS y SMA es que la ELA es una enfermedad de la neurona motora que parece esporádica la mayor parte del tiempo y rara vez se hereda, mientras que la SMA es una enfermedad de la neurona motora que siempre se hereda.
La esclerosis lateral amiotrófica (ELA) y la atrofia muscular espinal (SMA) son dos tipos diferentes de enfermedades de las neuronas motoras. Ambas enfermedades se caracterizan por una pérdida de neuronas motoras somáticas e inervaciones a los músculos esqueléticos voluntarios, lo que lleva a la muerte por fallas de la función respiratoria. ALS es principalmente esporádico y también se puede heredar, mientras que la SMA siempre es una enfermedad de neurona motora heredada. Además, la ELA se observa principalmente en adultos, mientras que la SMA se observa principalmente en niños y rara vez en adultos. Además, no hay una cura adecuada para ambas enfermedades en este momento.
CONTENIDO
1. Descripción general y diferencia de claves
2. Que es ALS
3. Que es SMA
4. Similitudes - ALS y SMA
5. Als vs sma en forma tabular
6. Resumen - ALS vs SMA
Que es ALS?
ALS es una enfermedad neurológica rara que afecta principalmente a los nervios responsables de controlar el movimiento muscular voluntario. Los músculos voluntarios son los músculos que la gente usa para moverse. ALS es la forma más común de la enfermedad de las neuronas motoras. Tiene un riesgo de por vida de 1: 300. ALS normalmente tiene un inicio de adultos y es muy agresivo. La mayoría de los pacientes mueren de 2 a tres años de diagnóstico. ALS parece esporádico en el 90% de los casos. Pero se puede heredar en el 10% de los casos.
Cuando se hereda, múltiples mutaciones en diferentes genes son agentes causales, como mutaciones en superóxido dismutasa 1 (Sod1) gen, Tardbp (TDP43) gen, Fusionado en el sarcoma (Fus) gen y Cromosoma 9 marco de lectura abierto 72 (C9ORF72) Gen. También se hereda en un patrón de herencia dominante autosómico. Los signos y síntomas de la ELA pueden incluir dificultad para caminar, tropezar y caer, debilidad a menudo en las piernas, los pies o los tobillos, la torpeza, el habla arrastrada, los problemas para tragar, los calambres musculares, la contracción en los brazos, los hombros y la lengua, el llanto, riendo y riendo y riendo Bostezando de manera inapropiada y los cambios en la función cognitiva y el comportamiento.
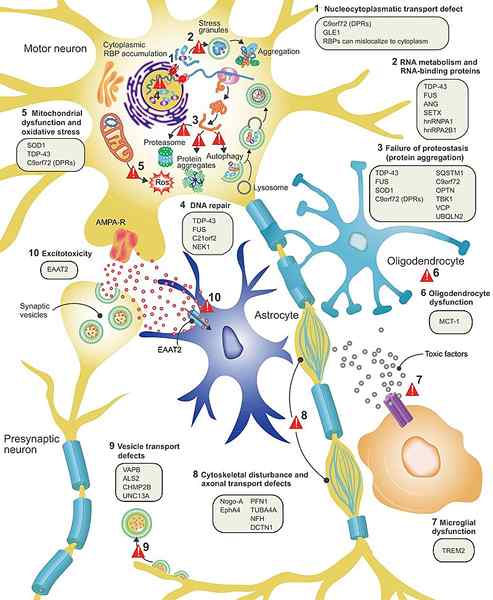
Figura 01: ALS
La esclerosis lateral amiotrófica se puede diagnosticar a través de un electromiograma (EMG), estudio de conducción nerviosa, IRM, estudio de sangre y orina, grifo espinal (punción lumbar) y biopsia muscular. Además, las opciones de tratamiento para la esclerosis lateral amiotrófica pueden incluir medicamentos como el riluzol, la edaravona, el fenilbutirato de sodio, el taurursodiol y las terapias como el cuidado respiratoria, la fisioterapia, la terapia ocupacional, la terapia del habla, la terapia nutricional, el apoyo psicológico y social.
Que es SMA?
La SMA es un trastorno hereditario de la neurona motora que destruye los nervios en el tallo cerebral y la médula espinal, que son esenciales para controlar las actividades del músculo esquelético como hablar, respirar y tragar. Esto finalmente conduce a la debilidad muscular y la atrofia. SMA generalmente muestra patrones de herencia autosómicos recesivos. El inicio de esta enfermedad ocurre antes de los 6 meses, y la letalidad a los 2 años de edad.
La SMA también es la causa genética más común de mortalidad infantil, con una incidencia anual de 1: 6000 a 1: 10000 nacimientos vivos. Además, la atrofia muscular espinal se debe a las mutaciones en el Survival Motor Neuron 1 (Smn1) gen, lo que conduce a bajos niveles de proteína SMN. Hay cuatro tipos de SMA llamados SMA1 (forma severa), SMA2 (forma intermedia), SMA2 (forma leve) y SMA4 (forma adulta). Los síntomas de la SMA pueden incluir un disquete o con los brazos y las piernas débiles, problemas de movimiento, temblar o temblores de músculos, problemas en huesos y articulaciones, problemas de deglución y dificultades para respirar.
La atrofia muscular espinal se puede diagnosticar mediante análisis de sangre, pruebas genéticas, pruebas de conducción nerviosa y biopsia muscular. Además, las opciones de tratamiento para la atrofia muscular espinal pueden incluir la terapia modificadora de la enfermedad (que dan a los medicamentos estimulantes de la producción de proteínas SMN como Nusinersen) y la terapia de reemplazo de genes (infusión intravenosa de un medicamento llamado Onasemnogene Abeparvovec-xioi que reemplaza o falla o falla o falla Smn1 gene).
¿Cuáles son las similitudes entre ALS y SMA??
- ALS y SMA son dos enfermedades de las neuronas motoras.
- Ambas enfermedades afectan principalmente el movimiento muscular voluntario.
- Son enfermedades raras.
- Ambas enfermedades pueden ser heredadas.
- Pueden causar letalidad severa.
- Ambas enfermedades se pueden identificar en adultos.
- Ambas enfermedades pueden tener síntomas similares, como problemas de movimiento, problemas de deglución y dificultades para respirar.
- Se pueden diagnosticar a través del examen físico, los análisis de sangre y la conducción nerviosa.
- Se tratan principalmente a través de medicamentos específicos.
¿Cuál es la diferencia entre ALS y SMA??
ALS es una enfermedad de las neuronas motoras que parece esporádica la mayor parte del tiempo y se hereda raramente, mientras que la SMA es una enfermedad de la neurona motora que siempre está heredada. Por lo tanto, esta es la diferencia clave entre ALS y SMA. Además, ALS muestra un patrón de herencia autosómico dominante cuando se hereda, mientras que la SMA muestra un patrón de herencia autosómico recesivo cuando.
La siguiente infografía presenta las diferencias entre ALS y SMA en forma tabular para la comparación de lado a lado.
Resumen -ALS vs SMA
Las enfermedades de las neuronas motoras son condiciones raras que dañan las partes del sistema nervioso progresivamente. ALS y SMA son los dos trastornos de la neurona motora más comunes. ALS es una enfermedad de las neuronas motoras que parece esporádica la mayor parte del tiempo y rara vez se hereda. Es autosómico dominante. La SMA es una enfermedad de la neurona motora que siempre se hereda. Es autosómico recesivo. Ambos pueden conducir a la muerte por insuficiencia respiratoria. Entonces, esta es la diferencia clave entre ALS y SMA.
Referencia:
1. “Esclerosis lateral amiotrófica (ELA)."Mayo Clinic, Mayo Foundation for Medical Education and Research.
2. "Descripción general: atrofia muscular espinal" NHS opciones, NHS.
Imagen de cortesía:
1. "Patología de la enfermedad de ELA y mecanismos propuestos de enfermedad" por Philip van Damme, Wim Robberecht y Ludo van den Bosch -"Modelado de esclerosis lateral amiotrófica: progreso y posibilidades" Modelos de enfermedades y mecanismos. 10 (5): 537-549. doi: 10.1242/dmm.029058 PMC: 5451175 PMID: 28468939 (CC por 3.0) a través de Commons Wikimedia